Zebrafish melanoma
You can download them from STMiner-test-data. You can also download the raw dataset from GEO.
Import package
from STMiner.SPFinder import SPFinder
Load data
file_path = 'Path/to/your/h5ad/file'
sp = SPFinder()
sp.read_h5ad(file=file_path, bin_size=1)
The parameter bin_size specifies the size of merged cells (spots). If not specified, no merging is performed. If set to 50, 50x50 cells/spots will be merged into a single cell/spot. Due to low sequencing depth in some datasets, cells/spots are often merged during analysis (e.g., stereo-seq). However, 10x data typically does not require merging.
The ST datasets was storaged in .adata object of sp, you can use sp.adata to check them:
sp.adata
Besides, sp Obj has many useful attributes which can be used for visualization or integrated into other pipelines (such as scanpy). See API for more details.
Find SVG
sp.get_genes_csr_array(min_cells=200, log1p=False)
sp.spatial_high_variable_genes(thread=6)
The parameter min_cells was used to filter genes that are too sparse to generate a reliable spatial distribution.
The parameter log1p was used to avoid extreme values affecting the results. For most open-source h5ad files, log1p has already been executed, so the default value here is False.
You can perform STMiner in your interested gene sets. Use parameter gene_list to input the gene list to STMiner. Then, STMiner will only calculate the given gene set of the dataset. You can see output while computing as follows:
Parsing distance array...: 100%|██████████| 10762/10762 [01:12<00:00, 149.11it/s]
Computing ot distances...: 10%|▉ | 1069/10762 [03:04<31:11, 6.12it/s]
You can check the spatial varitation of each gene by:
sp.global_distance
| Gene | Distance | z-score |
|---|---|---|
| myha | 1.35E+08 | 2.771493 |
| vmhcl | 1.01E+08 | 2.470881 |
| zgc:101560 | 9.95E+07 | 2.458787 |
| pvalb1 | 9.82E+07 | 2.445257 |
| myhz2 | 9.75E+07 | 2.437787 |
| ... | ... | ... |
| rps17 | 2.61E+05 | -3.63207 |
| rpl13 | 2.48E+05 | -3.68506 |
| rpl32 | 2.43E+05 | -3.70327 |
| rsl24d1 | 2.27E+05 | -3.7757 |
| rpl22 | 1.83E+05 | -3.99332 |
Fit GMM
sp.fit_pattern(n_comp=20, gene_list=list(sp.global_distance[:2000]['Gene']))
You can see output while computing as follows:
Fitting GMM...: 10%|▉ | 190/2000 [00:42<04:36, 6.54it/s]
n_comp: Number of components for each GMM model
gene_list: Gene list to fit GMM model
Build distance array
sp.build_distance_array()
This step calculates the distance between genes’ spatial distributions. You can visualize the distance array by:
import seaborn as sns
sns.clustermap(sp.genes_distance_array)
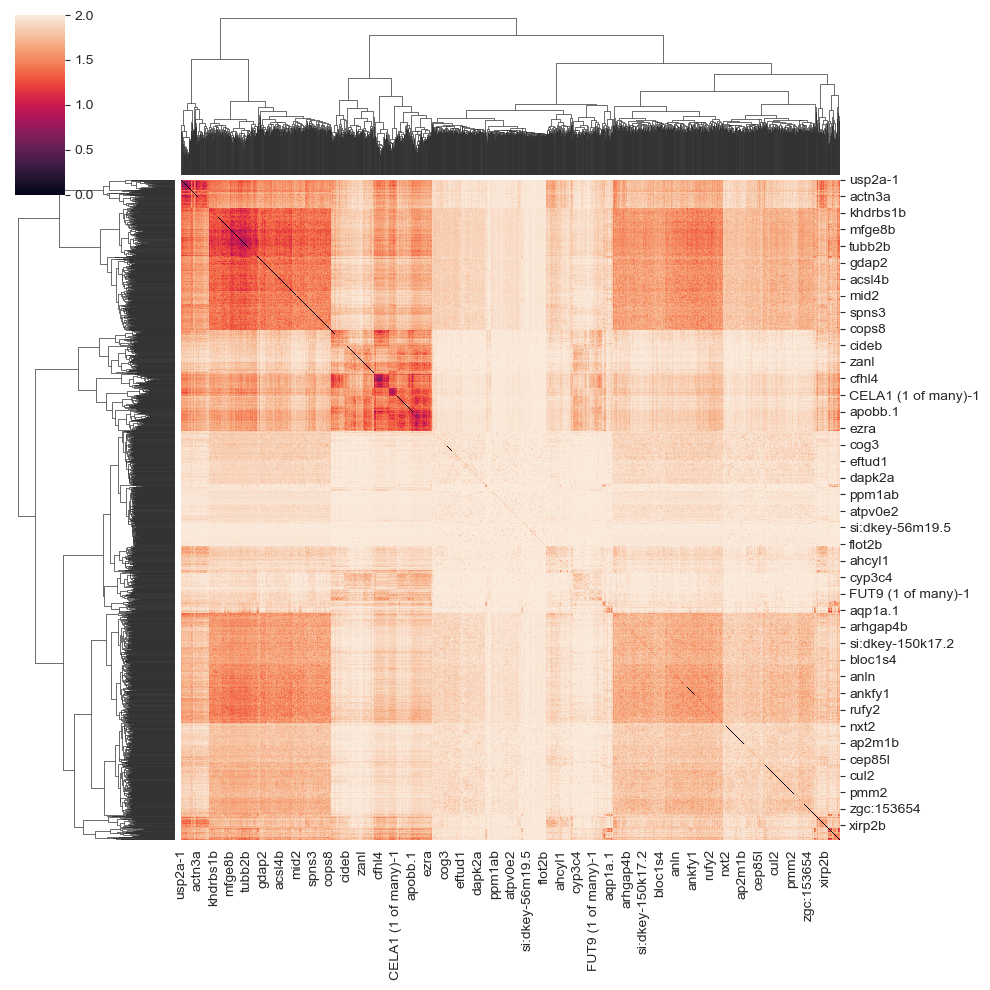
build distance matrix & clustering
sp.cluster(n_clusters=6)
n_clusters: Number of cluster
Result & Visualization
The result are stored in genes_labels:
spf.genes_labels
The output looks like the following:
| gene_id | labels | |
|---|---|---|
| 0 | Cldn5 | 2 |
| 1 | Fyco1 | 2 |
| 2 | Pmepa1 | 2 |
| 3 | Arhgap5 | 0 |
| 4 | Apc | 5 |
| .. | ... | ... |
| 95 | Cyp2a5 | 0 |
| 96 | X5730403I07Rik | 0 |
| 97 | Ltbp2 | 2 |
| 98 | Rbp4 | 4 |
| 99 | Hist1h1e | 4 |
To visualize the patterns by heatmap:
sp.get_pattern_array()
sp.plot.plot_pattern(heatmap=False,
s=5,
rotate=True,
reverse_y=True,
reverse_x=True,
vmax=95,
cmap='Spectral_r',
output_path='./')
heatmap: If True, plot a heatmap. If False, plot a scatterplot. False is the default.
s: Spot size
rotate\reverse_y\reverse_x: Adjust the axis of plot.
cmap: cmap of plot
vmax: The percentage of the highest value of plots. Avoid the effect of large values for visualization.
output_path: If set, save the figure to path To visualize the genes by labels:
sp.plot.plot_genes(label=0, n_gene=8, s=5, reverse_y=True, reverse_x=True)
n_gene: Number of genes to visualize
To visualize the specific gene (such as BRAFhuman):
hcc2l.plot.plot_gene('BRAFhuman',
spot_size=10,
global_matrix_spot_size=10,
rotate=True,
reverse_y=True,
reverse_x=True,
vmax=95,
cmap='Spectral_r',
figsize=(5,5),
save_path='./',
format='png')
reverse_y, reverse_x, rotate is optional, they are used to adjust coordinate here.